02 Jun 2026
The final is NOT comprehensive, it will focus on topics since the midterm but because the class is cumulative, there may be some questions that require you to apply concepts from earlier in the class.
The final will be in two parts: a take-home part and an in-class part. The take-home part will be open book/web and unlimited time. The in-class part will be closed book but you can bring one hand-written page of notes.
In Class part
This will be a concept and knowledge-based exam and will not include true scripting/coding. You may be asked to pseudo-code or to find a code error.
You can bring one hand-written page of notes.
Study guide
- Know the purpose of the the various file formats we have used (fastq, sam/bam, vcf) and what types of data they contain. I will not ask you to recreate one of these but I likely will give you a snippet of one and ask you to explain/interpret the columns.
- Know what a PHRED score is and how it relates to the probability of error in a base call
- Be able to convert a PHRED score into a probability of error and vice versa. You should be able to do this without a calculator for simple scores (e.g. 10, 20, 30) and you should know the formula for converting between the two so that you could do it with a calculator for more complex scores.
- Know the difference between PHRED+33 and PHRED+64. You do not need to memorize the ASCII codes for the characters, but if I gave you the PHRED+33 and PHRED+64 character keys along with a fastq file snippet, you should be able to determine which PHRED encoding is being used and convert the quality scores to probabilities of error.
- What is mapping quality and how does it differ from base quality?
- How to read a CIGAR string and what the various indicators (M, N, I, D) mean.
- I will NOT ask you to interpret the bitwise FLAG.
- How can you (or a SNP caller) distinguish between a true SNP and a sequencing error?
- What are the steps involved between getting raw sequencing data and performing differential gene expression analysis? What are the tools we have used for each step and what do they do?
- What is clustering, why would we want to do it? What methods have we used for clustering and how do they differ? (Hierarchical vs k-means)
- Know the steps for constructing a gene co-expression network.
- Define node, edge, and degree in the context of a gene co-expression network.
- Explain the difference between degree and betweenness centrality in a gene co-expression network and how they can be used to identify important genes in the network.
- Given a simple network graph, be able to pick the node with the highest degree and the node with the highest betweenness centrality.
- What is the basis for GO and promoter enrichment analyses?
- Know the difference between alpha and beta diversity and what they are used for in the context of metagenomics.
Example Questions
Question 1
Consider the following data from an Illumina sequencing experiment:
A00887:346:H2VK2DSX2:1:1141:3884:31986 163 scaffold_0 43675 255 70M89N73M = 43736 293
CATTTCTCACCTCCTCAAGGCAACTTTCAAGCTCCTTCAATTCTTCATCCTCCGAGAAGCTCACTGTGGCTTGTTTGATTGTGTTCTTCAAATGCATCTCAGCACTAAAGAGCTCTCGCCTGCTTCCTGTGGACACTGAGATC
FI,5FFFFFFFFFFFFFFFFFFF,FFFFFF:FFFFFF,FF:FFFFF:FFFFFFFFFFF:FFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFF,FF:FFFFFFFFFFFFFF:
NH:i:1 HI:i:1 AS:i:276 nM:i:6
A (2pts): The data above comes from what kind of file?
B: (3pts): The sequencing quality is in Phred+33 or Phred+64?
(Note: I would give you this chart for the exam.)
C: (2pts) Convert the quality of the fourth base to a Phred score.
D: (2pts) Convert the Phred score from part C to a p-value. If you aren’t sure how to do this, give the formula.
E: (3 pts) Explain what the probability from D means in terms of confidence in the sequence. If you are stuck on C or D then just pick a p-value to illustrate (like what does a p-value of 0.01 mean in this context).
F: (2pts) What does the number “43675” refer to?
G: (2pts) What does the number “255” refer to?
H: (2pts) What does “70M89N73M” mean?
Question 2
You have performed a differential gene expression analysis to find genes expressed differentially in the developing petals of red vs white roses. You have a list of 963 genes that are higher in red petals and 1204 genes that are higher in white petals. Briefly describe three follow-up analyses you could do with these gene lists to try to understand the biological basis for the differences in petal color. For each analysis explain what you would hope to learn. Two sentences for each analysis.
Question 3
Examine the gene co-expression network graph below.
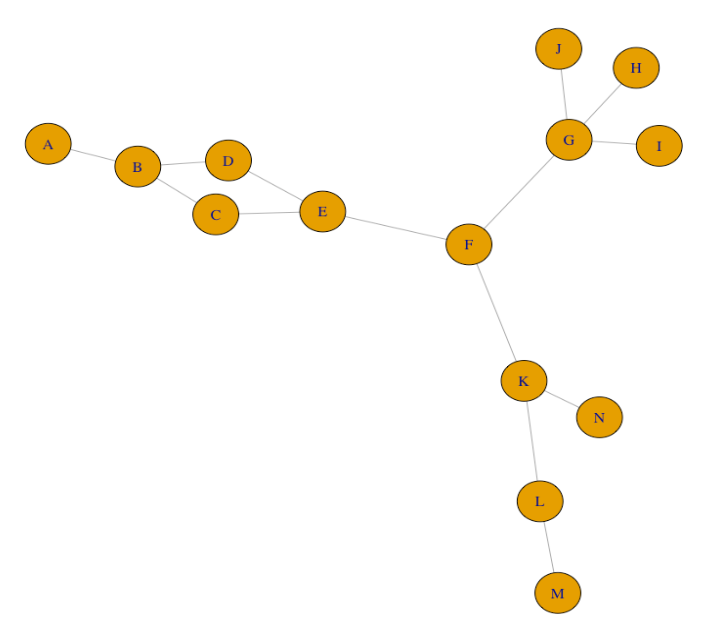
A: What is a node in this graph? Describe what the nodes in this graph represent.
B: What is an edge in this graph? Describe what the edges in this graph represent.
C: Which gene has the highest degree centrality? Explain your choice.
D: Which gene has the highest betweenness centrality? Explain your choice.
Question 4
You are examining a .vcf file. On different rows you see the following entries. What does each of these mean?
A: 0/1
B: 1/1
C: 0/0
Question 5 You have performed a metagenomics analysis of the microbial communities in the soil of a tomato farm and the soil of a nearby grassland. You find that the alpha diversity of the tomato farm soil is much lower than that of the grassland soil. What does this mean? What could be some reasons for this difference?
Note: not all topics covered in the final will be represented in the example questions above. The example questions are meant to give you a sense of the types of questions that will be on the exam, but they are not meant to be an exhaustive list of topics or question types.
Take Home
No study guide for this; it will be open book/web and unlimited time. You will be given data sets and asked to do analyses similar to what you have done in the labs and assignments.
23 Apr 2026
The midterm will be in two parts.
Part one will be in class and closed book, but You can bring one hand-written page of notes.
Part two will be take home and open book.
Part One–In Class
This will be a concept and knowledge-based exam and will not include true scripting/coding. You may be asked to pseudo-code or to find a code error.
You can bring one hand-written page of notes.
Study guide
- Know the general format of Linux and R commands.
- Be able to explain / interpret and create relative and absolute file paths.
- Know which Linux commands you would use to
- rename a file
- copy a file
- move a file
- navigate the file system
- look at file size
- see the contents of a file
- Know how to create and access variables in Linux. What does
$ do in Linux?
- What does
$(CMD) do in Linux? (Where CMD is a Linux command).
- Know the difference between
git add, git commit, git pull and git push.
- Know the structure of a
for loop in Linux. Be able to pseudo-code a for loop.
- BLAST
- What is the format of a FASTA file?
- What are the different BLAST programs and how do you choose?
- What is word size, and how do you choose?
- What is an e-value and how do you interpret it?
- Be able to interpret a PCA plot of SNP data.
- How can you tell if population structure is a problem for your trait?
- Be able to interpret a phylogenetic tree
- Explain how information is passed from the UI to the server in a Shiny app.
- How does GWAS work?
- What is a QQ plot? be able to interpret one.
- What is population structure and how does it affect GWAS?
- Be able to interpret a Manhattan plot
- Genome Assembly
- What does BUSCO stand for, and how is it used to evaluate genome assemblies?
- What is N50 and how is it calculated? How is it used to evaluate genome assemblies?
Example Questions
1A: You have cloned a gene of interest from the Eastern Monarch Butterfly population and you want to find its ortholog in Western Butterfly via a nucleotide BLAST search. You expect the sequences to be very similar. Considering the word sizes we looked at in the BLAST lab, what word size you use? Why? (Conceptual question, no code needed)
1B: Will your choice in part 1A cause an increased or decreased sensitivity (ability to detect distant homologs)? Why? (Conceptual question, no code needed)
1C: If you wanted to blast all of the genes in the Eastern Monarch Butterfly genome against those in the Western Monarch Butterfly genome to find close orthologs would that change your word size choice?
2 You want to rename the file monarch_genome.fasta to monarch_genome_v1.fasta. What Linux command would you use? Include the command and how the file names would be used in the command.
3 what git commands would you use to update your local repository with the latest changes from the remote repository, and then to submit your changes to the remote repository?
4 Compare and contrast git pull and git clone: how are these related and what makes them different?
5 You have 100 FASTA files in a directory. You want to run a separate BLAST search on each of them. Write a pseudo-code for loop that would accomplish this task. You do not need to write the actual BLAST command, just the structure of the loop and how you would use the file names in the loop.
6 The code below does not work. What is the error?
testFiles=$(ls test*.txt)
for file in $testFiles
do
cat file
done
The output is
cat: file: No such file or directory
cat: file: No such file or directory
cat: file: No such file or directory
Note: not all topics covered in the midterm will be represented in the example questions above. The example questions are meant to give you a sense of the types of questions that will be on the exam, but they are not meant to be an exhaustive list of topics or question types.
Part Two–Take Home
No study guide for this; it will be open book/web and unlimited time. You will be given data sets and asked to do analyses similar to what you have done in the labs and assignments.
21 Feb 2026
Dear Students,
Welcome to BIS180L. I am looking forward to teaching you in this class. This class will give you hands-on experience with bioinformatics data analysis.
Class will meet in person in TLC 2216
Pre-recorded Lectures
For the most part I will provided lectures in a pre-recorded format. This will allow the best use of our time together in the classroom. Pre-recorded lectures will be available at the latest at 9AM the day before the lecture/lab time. You will need to watch them in advance and answer embedded quiz questions via playposit. Due date for the quiz is 9AM the day of lecture. This give me and the TA time to review questions before class.
There is no video due for the March 31 class
Class meeting time
Even though lectures are pre-recorded, we will still meet at 1:10 PM.
In class
We will devote the beginning of each class / lab to questions on the lecture material. You will then work in small groups of two or three to work on the lab material. John (TA) and I will be available to help when you have questions.
I expect you to be in class/lab each day at 1:10. If you have a Covid or other health emergency that prevents you from coming to class, please let me know (in advance if possible).
Outside of class
You can expect to spend a fair bit of time outside of class working on the assignments. This can either be done in one of the computer labs, or on your laptop. If you are not willing to put in time outside of class this is probably not the best class for you. If you do put in the time outside of class you will be rewarded by learning a great deal about how to perform bioinformatics analyses.
21 Feb 2026
We will use Vince Buffalo’s excellent book Bioinformatics Data Skills.
This book is available online for free through the UC Davis Library (on campus or VPN connection required). UC is licensed for simultaneous access by 28 people. If that doesn’t work for you, you can buy it direct from the publisher or from amazon. As of this posting Amazon is cheaper and has used copies as well.
Additionally we will use Hadley Wickham’s also excellent book R for Data Science which is available online for free. If you would like a physical copy, here is the amazon link
{kind=link}